It is the time of the year again and we are running our advent calendar again. The first door opened and reveals an update on the Biodiversity Genomics Europe (BGE) project. The project started about a year ago and it is time to take a first look at the accomplishments of the project. The European Reference Genome Atlas (ERGA) stream of the project also contains a task called community sequencing, which is led by us at UiO. We are not sequencing our community, but the community can suggest species, whose genome shall be sequenced. To select from these species, together with the Sampling and Sample Processing (SSP) committee we developed an automated selection procedure based on objective criteria and without human intervention in the selection process. After two calls, we have now selected about 100 species across the eukaryotic Tree of Life totaling a genome size of 150 Gb. The sample providers are now busy sampling the species and sending them to the sequencing centers of the project.
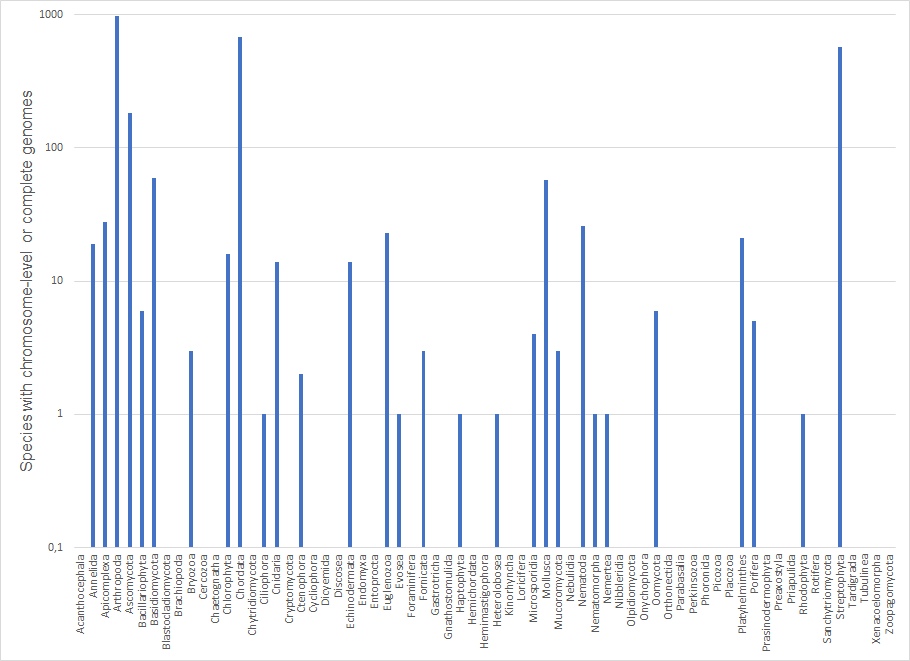
We also looked at the distribution of available genomes per phylum, which also showed that we need a better taxonomic representation of reference genomes across the eukaryotic tree of life. As of July 28th 2023, of the 67 eukaryotic phyla listed in NCBI (National Center for Biotechnology Information) only 29 were represented in GoaT by at least one species with a chromosome-level or complete genome, while 38 phyla lacked a reference genome (e.g., in the animals: Brachiopoda, Chaetognatha, Cycliophora, Dicyemida, Entoprocta, Gastrotricha, Gnathostomulida, Hemichordata, Kinorhyncha, Loricifera, Onychophora, Orthonectida, Phoronida, Placozoa, Priapulida, Rotifera, Tardigrada, and Xenacoelomorpha). Among the sequenced phyla, the number of sequenced genomes per phylum was highly uneven with 15 phyla having fewer than 10 species with chromosome-level or complete genomes, 10 having between 10 and 100 species with such genomes, and four having more than 100 species with such genomes. Without a better taxonomic representation in reference genome generation, we are facing huge knowledge gaps in our understanding of the evolution and ecology of biodiversity on Earth and thus lack knowledge on how to protect and preserve it.
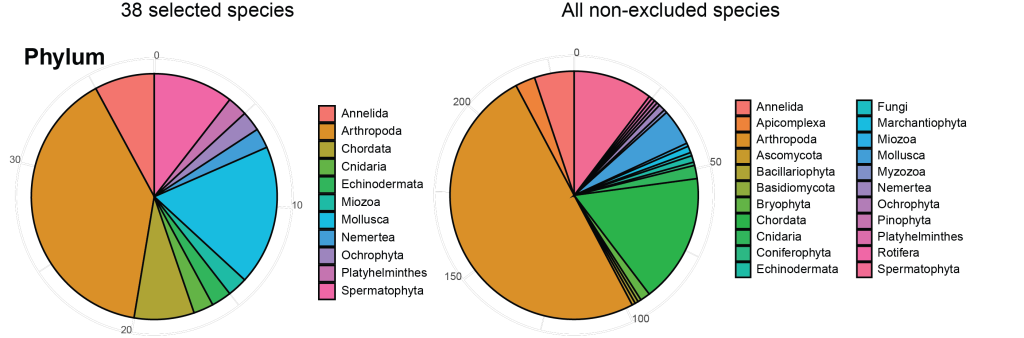
The selection process also allows us to take a closer investigation of the selected species and what affected their selection the most. The selected 38 species after the first call comprise 11 phyla of the originally 22 suggested phyla. Most of them, 10 out of 11, were excluded due to the feasibility check. In relative terms arthropods, chordates and spermatophytes are less represented among the selected than across all species, while annelids and molluscs are stronger represented.
The feasibility check generally has a very strong effect on the species selection in both rounds. In the first call, 120 of 230 species were considered feasible. This means that almost half (47.8%) of the suggested species that were considered not feasible. Closer examination revealed the following reasons. Of the non-feasible species, 18.2% were excluded due to a genome size of larger than 6 Gb and 28.2% due to a sample body size smaller than a Drosophila fly or less than 100,000 nucleated cells. 44.5% of the species were excluded because they fulfilled one the two criteria or both. 1.8% of the species have a small body size with large genomes. All of these species are challenging even for new sequencing technologies and hence new methods are needed to be developed. Many of these species are meiofauna species, which are generally obtain less attention in research.
On the other hand, 55.5% of the species were considered not feasible due to criteria related purely to the sampling and sample processing. 22.7% of the species could not be snap-frozen or frozen on dry ice for sample preservation. For 37.3% of the non-feasible species, it was not possible to preserve them within 5 minutes of their death and for 28.2% it was not possible to maintain a strict cold chain at -70°C. Please keep in mind that a single species could not be feasible due to more than criterion. For example, 6.4% of the species have both a too small body size and could not be preserved within 5 minutes upon death. The challenges of collecting meiofauna species suitable for genomic research is described in a recent blog and how a masonry trowel is used in genomic research.
Finally, 18% of the species did not fulfill the criterion that they were already collected or easy to obtain. Of these, 55% were also regarded as non-feasible due to the other criteria, but 45% of them or 8.2% of all non-feasible species are just challenging to collect. Accordingly, 47.3% of the non-feasible species were excluded because they could not properly be preserved or maintained. Hence, besides an improvement in our methodology we also need to improve our logistics.
![]()
1 Comment on “Door 1: Sequencing genomes suggested by the community”