
As the nights get longer and the end of the year draws ever nearer, the FEZ advent calendar gives us the opportunity to introduce what we’ve been working on this year to everyone. My role in the group is tied to the Invertomics project, the quest for a better understanding of invertebrate genetics and genomics, but this year, I’ve not really been working on any invertebrate data at all!
As a postdoctoral researcher and the group’s designated “methods person”, I’ve been looking at why the invertebrate tree of life has been so difficult to resolve in the first place. That means looking at all the previously published data, and the competing hypotheses of different groups as to how all these different animals are related; and working out the root cause of all these disagreements.
Whilst other members of the group are out gathering new data and working hard in the lab, I’m approaching our problem from the other direction, trying to work out new ways to evaluate how we understand evolution as a process, and how we make models that simulate it.
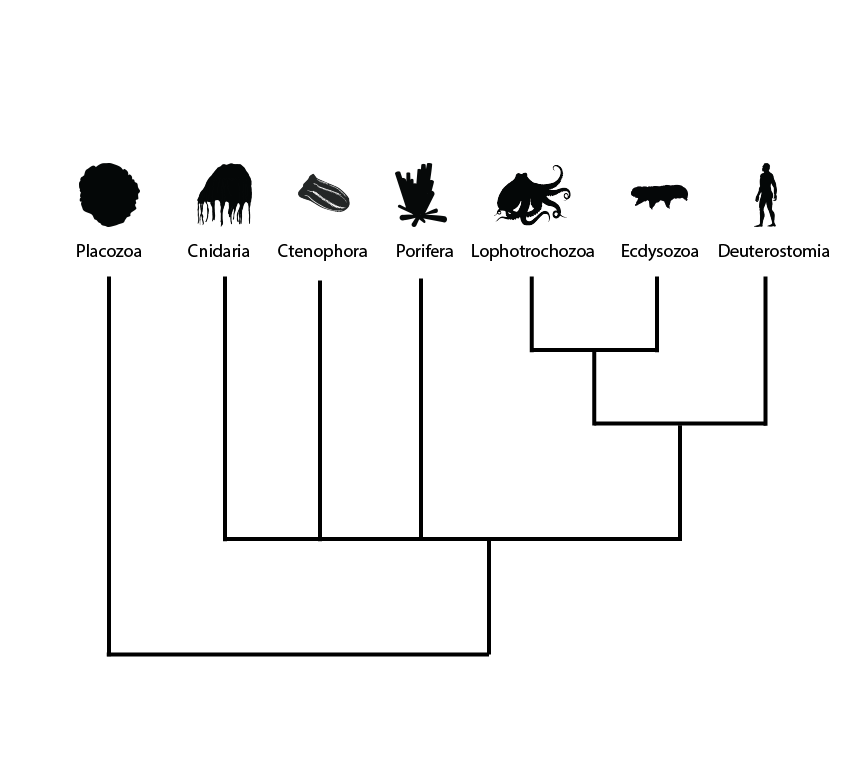
When we make phylogenies (pictured) – trees of relationships – from genes, what we are really doing is a multi-step process. First, we identify the gene in the genome. Then, we identify the same gene across lots of different organisms and put them all together, trying to match up the same bits of the gene against one another, called an alignment (Cover Image pictured). We use this to then build a tree, by taking a model that matches how evolution could work – by changing parts of the gene from one nucleic acid to another through mutation, or by removing old parts and adding new parts by deletion and insertion. This model traces back from each of the sequences, working out the arrangement of all of them that most makes sense, given how likely the model thinks certain changes are over time.
Our models are good, but they aren’t perfect. And that is where my job comes in. As approximations of evolution, these models can be susceptible to a lot of errors: some because the reality of evolution is more complex than we previously anticipated or can accurately model, and some because the data we have is not yet perfect. This year I’ve been working on a new measurement, called nRCFV, which might help us understand and identify one of the biggest problems in accurately reconstructing these trees – compositional heterogeneity (a very scientific name for “this gene, or part of a gene, has changed so much that it is really different from all the others of the same type”). I’ve also been working on a new kind of reliability measurement to test our hypotheses of these relationships, which will help us evaluate how many genes support different hypotheses.
Biology can sometimes seem like a science done with a lab coat or out in the field, but there’s a lot of work still to be done in the server rooms and behind computers to help us understand how life really works!
![]()